第三章 紫外-可见分光光度法
Ultraviolet and visible
spectrophotometry,UV-Vis
紫外-可见分光光度法是利用某些物质的分子吸收200~800nm光谱区的辐射来进行分析测定的方法。这种分子吸收光谱产生于价电子和分子轨道上的电子在电子能级间的跃迁,广泛用于无机和有机物质的定性和定量测定。
3.1 紫外-可见吸收光谱
分子中的电子总是处在某一种运动状态中,每一种状态都具有一定的能量,属于一定的能级。电子由于受到光、热、电等的激发,从一个能级转移到另一个能级,称为跃迁。当这些电子吸收了外来辐射的能量,就从一个能量较低的能级跃迁到另一个能量较高的能级。但是由于分子内部运动所牵涉到的能级变化比较复杂,分子吸收光谱也就比较复杂。在分子内部除了电子运动状态外,还有核间的相对运动,即核的振动和分子绕着重心的转动。而振动能和转动能,按量子力学计算,它们是不连续的,即具有量子化的性质。所以,一个分子吸收了外来辐射之后,它的能量变化![]() 为其振动能变化
为其振动能变化![]() 、转动能变化
、转动能变化![]() ,以及电子运动能量变化
,以及电子运动能量变化![]() 之总和,即
之总和,即
![]() (3.1)
(3.1)
若分子的较高能级与较低能级能量之差恰好等于电磁波的能量![]() 时,有
时,有
![]() (3.2)
(3.2)
则分子将从较低能级跃迁到较高能级。式(3.1)中![]() 最大,一般在1~20eV之间。现假设其为5eV,由式(3.2)计算出其相应的波长为250nm。因此,由分子内部电子能级的跃迁而产生的光谱位于紫外-可见光区内。
最大,一般在1~20eV之间。现假设其为5eV,由式(3.2)计算出其相应的波长为250nm。因此,由分子内部电子能级的跃迁而产生的光谱位于紫外-可见光区内。
分子的振动能级间隔![]() 大约比
大约比![]() 小10倍,一般在0.05~1eV之间。如果
小10倍,一般在0.05~1eV之间。如果![]() 为0.1eV,即为5eV的电子能级间隔的2%。因此在发生电子能级之间跃迁的同时,必然也要发生振动能级之间的跃迁,得到的是一系列的谱线,它们相互波长的间隔是250nm×2%=5nm,而不是250nm单一的谱线。
为0.1eV,即为5eV的电子能级间隔的2%。因此在发生电子能级之间跃迁的同时,必然也要发生振动能级之间的跃迁,得到的是一系列的谱线,它们相互波长的间隔是250nm×2%=5nm,而不是250nm单一的谱线。
分子的转动能级间隔![]() 大约比
大约比![]() 小10倍或100倍,一般小于0.05eV。现假设
小10倍或100倍,一般小于0.05eV。现假设![]() 为0.005eV,则为5eV的电子能级间隔的0.1%。当发生电子能级和振动能级之间的跃迁时,必然也要发生转动能级之间的跃迁。由于得到的谱线彼此间的波长间隔只有250nm×0.1%=0.25nm,如此小的间隔使它们连在一起,呈现带状,称为带状光谱(band
spectrum)。
为0.005eV,则为5eV的电子能级间隔的0.1%。当发生电子能级和振动能级之间的跃迁时,必然也要发生转动能级之间的跃迁。由于得到的谱线彼此间的波长间隔只有250nm×0.1%=0.25nm,如此小的间隔使它们连在一起,呈现带状,称为带状光谱(band
spectrum)。
图3.1是双原子分子的能级示意图,图中![]() 和
和![]() 表示不同能量的电子能级,在每个电子能级中因振动能量不同而分为若干个v =0,1,2,3…的振动能级,在同一电子能级和同一振动能级中,还因转动能量不同而分为若干个
表示不同能量的电子能级,在每个电子能级中因振动能量不同而分为若干个v =0,1,2,3…的振动能级,在同一电子能级和同一振动能级中,还因转动能量不同而分为若干个![]() =0,1,2,3……的转动能级。
=0,1,2,3……的转动能级。
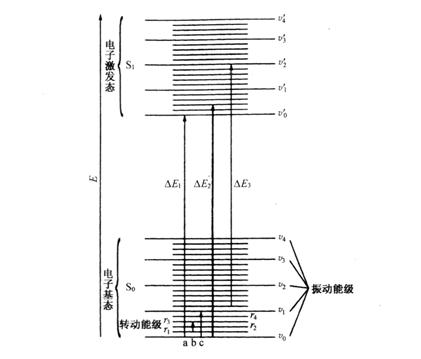
图3.1 分子能级和电子能级跃迁
(a)转动能级跃迁 (b)振动能级跃迁(c)
物质对不同波长的光线具有不同的吸收能力,物质也只能选择性地吸收那些能量相当于该分子振动能变化![]() 、转动能变化
、转动能变化![]() 以及电子运动能量变化
以及电子运动能量变化![]() 的总和辐射。由于各种物质分子内部结构的不同,分子的能级也是千差万别,各种能级之间的间隔也互不相同,这样就决定了它们对不同波长光线的选择吸收。如果改变通过某一吸收物质的入射光的波长,并记录该物质在每一波长处的吸光度(
的总和辐射。由于各种物质分子内部结构的不同,分子的能级也是千差万别,各种能级之间的间隔也互不相同,这样就决定了它们对不同波长光线的选择吸收。如果改变通过某一吸收物质的入射光的波长,并记录该物质在每一波长处的吸光度(![]() ),然后以波长为横坐标、以吸光度为纵坐标作图,这样得到的谱图称为该物质的吸收光谱或吸收曲线。某物质的吸收光谱反映了它在不同的光谱区域内吸收能力的分布情况,可以从波形、波峰的强度、位置及其数目看出来,为研究物质的内部结构提供重要信息。
),然后以波长为横坐标、以吸光度为纵坐标作图,这样得到的谱图称为该物质的吸收光谱或吸收曲线。某物质的吸收光谱反映了它在不同的光谱区域内吸收能力的分布情况,可以从波形、波峰的强度、位置及其数目看出来,为研究物质的内部结构提供重要信息。
有机化合物的紫外-可见光谱决定于分子的结构以及分子轨道上电子的性质。有机化合物分子对紫外光或可见光的特征吸收,可以用最大吸收处的波长,即吸收峰波长来表示,符号为![]() ,
,![]() 决定于分子的激发态与基态之间的能量差。从化学键的性质来看,与紫外-可见光谱有关的电子主要有三种,即形成单键的
决定于分子的激发态与基态之间的能量差。从化学键的性质来看,与紫外-可见光谱有关的电子主要有三种,即形成单键的![]() 电子、形成双键的
电子、形成双键的![]() 电子以及未参与成键的n电子(孤对电子)。
电子以及未参与成键的n电子(孤对电子)。
根据分子轨道理论,分子中这三种电子的能级高低次序是
(![]() )<(
)<(![]() )<(n)<(
)<(n)<(![]() )<(
)<(![]() )
)
![]() 、
、![]() 表示成键分子轨道;n表示非键分子轨道;
表示成键分子轨道;n表示非键分子轨道;![]() 、
、![]() 表示反键分子轨道。
表示反键分子轨道。![]() 轨道和
轨道和![]() 轨道是由原来属于原子的s电子和
轨道是由原来属于原子的s电子和![]() 电子所构成,
电子所构成,![]() 轨道
轨道![]() 轨道是由原来属于原子的
轨道是由原来属于原子的![]() 和pZ子所构成,n轨道是由原子中未参与成键的p电子所构成。当受到外来辐射的激发时,处在较低能级的电子就跃迁到较高的能级。由于各个分子轨道之间的能量差不同,因此要实现各种不同的跃迁所需要吸收的外来辐射的能量也是各不相同。有机化合物分子常见的4种跃迁类型是:
和pZ子所构成,n轨道是由原子中未参与成键的p电子所构成。当受到外来辐射的激发时,处在较低能级的电子就跃迁到较高的能级。由于各个分子轨道之间的能量差不同,因此要实现各种不同的跃迁所需要吸收的外来辐射的能量也是各不相同。有机化合物分子常见的4种跃迁类型是:![]() ,
,![]() ,
,![]() 和
和![]() 电子跃迁时吸收能量的大小顺序表示为
电子跃迁时吸收能量的大小顺序表示为
![]() >
>![]() >
>![]() >
>![]()
图3.2定性地表示了几种分子轨道能量的相对大小及不同类型的电子跃迁所需吸收能量的大小。
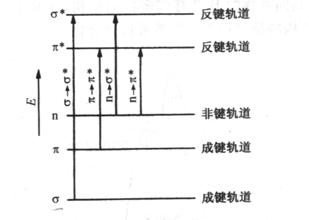
图3.2 分子的电子能级
1.饱和有机化合物
饱和烃分子中只有C—C键和C—H键,只能发生![]() 跃迁,其跃迁所需吸收的能量最大,因而所吸收的辐射波长最短,处于小于200nm的真空紫外区。如甲烷的
跃迁,其跃迁所需吸收的能量最大,因而所吸收的辐射波长最短,处于小于200nm的真空紫外区。如甲烷的![]() 为125nm,乙烷的
为125nm,乙烷的![]() 为135nm。
为135nm。
如果饱和烃中的氢原子被氧、氮、卤素等原子或基团所取代,这些原子中含有n电子,可以发生![]() 跃迁,其吸收峰有的在200nm附近,但大多数仍出现在小于200nm的区域内,
跃迁,其吸收峰有的在200nm附近,但大多数仍出现在小于200nm的区域内,![]() 跃迁的摩尔吸光系数
跃迁的摩尔吸光系数![]() 一般在100~
一般在100~
2.不饱受和脂肪族化合物
(1)![]() 跃迁 如含C=C、C=C或C=N键的分子能发生这一类电子跃迁,其特征是
跃迁 如含C=C、C=C或C=N键的分子能发生这一类电子跃迁,其特征是![]() 较大,一般在5×103~
较大,一般在5×103~![]() 跃迁一般在200nm左右,但具有共同轭双键的化合物,随着共轭体系的延长,
跃迁一般在200nm左右,但具有共同轭双键的化合物,随着共轭体系的延长,![]() 跃迁的吸收带将明显向长波方向移动,吸收强度也随之增强(见表3.1)。
跃迁的吸收带将明显向长波方向移动,吸收强度也随之增强(见表3.1)。
表3.1 多烯化合物的吸收带
|
化合物 |
双键数 |
|
颜色 |
|
乙烯 |
1 |
185(10 000) |
无色 |
|
丁二烯 |
2 |
217(21 000) |
无色 |
|
1,3,5-己三烯 |
3 |
258(35 000) |
无色 |
|
癸五烯 |
5 |
335(118 000) |
淡黄 |
|
二氢- |
8 |
415(210 000) |
橙黄 |
|
番茄红素 |
11 |
470(185 000) |
红 |
(2)n→![]() 跃迁 如含—OH、—NH2、—X、—S等基团的不饱和有机化合物,除了进行
跃迁 如含—OH、—NH2、—X、—S等基团的不饱和有机化合物,除了进行![]() →
→![]() 跃迁外,其杂原子中的孤对电子还可以发生
跃迁外,其杂原子中的孤对电子还可以发生![]() 跃迁,一般发生在近紫外区,吸收强度弱,
跃迁,一般发生在近紫外区,吸收强度弱,![]() 为10~
为10~
3.芳香族化合物
芳香族化合物一般都有E1带、E2带和B带3个吸收峰。苯蒸气的E1带![]() =184nm(
=184nm(![]() =4.7×104),E2带
=4.7×104),E2带![]() =204nm(
=204nm(![]() =6900),B带
=6900),B带![]() =255nm(
=255nm(![]() =230nm)(见图3.3)。在气态或非极性溶剂中,苯及其同系物的B带有许多精细结构,这是由于振动跃迁在基态电子跃迁上的叠加。这种精细结构特征可用于鉴别芳香族化合物。
=230nm)(见图3.3)。在气态或非极性溶剂中,苯及其同系物的B带有许多精细结构,这是由于振动跃迁在基态电子跃迁上的叠加。这种精细结构特征可用于鉴别芳香族化合物。
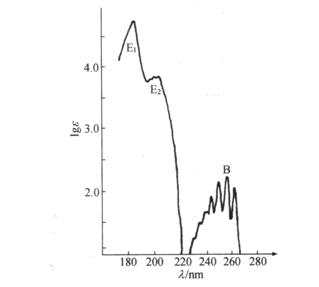
图3.3 苯在蒸气相中的紫外吸收光谱
对于稠环芳烃,随着苯环数目的增多,E1、E2和B带三个吸收带均向长波方向移动。
1.电荷转移光谱
某些分子同时具有电子给予体部分和电子接受体部分,它们在外来辐射激发下会强烈吸收紫外光或可见光,使电子从给予体外层轨道向接受体跃迁,这样产生的光谱称为电荷转移光谱(charge-transfer spectrum)。许多无机配合物能产生这种光谱。如以M和L分别表示配合物的中心离子和配位体,当一个电子由配位体的轨道跃迁到与中心离子相关的轨道上时,可用
下式表示:
![]() Mn+ — Lb- hν M(n-1)+ —L(b-1)-
Mn+ — Lb- hν M(n-1)+ —L(b-1)-
![]() 例如:
Fe3+ — SCN- hν Fe2+ —SCN
例如:
Fe3+ — SCN- hν Fe2+ —SCN
(接受体) (给予体)
一般来说,在配合物的电荷转移过程中,金属离子是电子接受体,配位体是电子给予体。此外,一些具有d10电子结构的过渡元素形成的卤化物及硫化物,如AgBr、PbI2、HgS 等,也是由于这类电荷转移而产生颜色。
有些有机化合物也可以产生电荷转移光谱。如在![]() 分子中,苯环可以作为电子给予体,氧可以作为电子接受体,在光子的作用下产生电荷转移:
分子中,苯环可以作为电子给予体,氧可以作为电子接受体,在光子的作用下产生电荷转移:
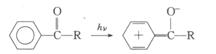
又如在乙醇介质中,将醌与氢醌混合产生暗绿色的分子配合物,它的吸收峰在可见光区。
电荷转移光谱谱带的最大特点是摩尔吸光系数大,![]() >
>
2.配位体场吸收光谱
配位体场吸收光谱(ligand
field absorption spectrum)是指过渡金属离子与配位体(通常是有机化合物)所形成的配合物在外来辐射作用下,吸收紫外或可见光而得到相应的吸收光谱。元素周期表中第4、第5周期的过渡元素分别含有3d和4d轨道,镧系和锕系元素分别含有
图3.4为在八面体场中d轨道的分裂示意图。由于它们的基态与激发态之间的能量差别不大,这类光谱一般位于可见光区。又由于选择规则的限制,配位场跃迁吸收谱带的摩尔吸光系数较小,一般![]() <
<
析中,但它可用于研究配合物的结构及无机配合物键合理论等方面
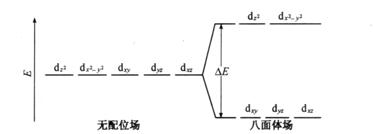
图3.4 在八面体场中d轨道的分裂
1.吸收光谱
吸收光谱(absorption
spectrum)又称吸收曲线,是以波长![]() (nm)为横坐标、以吸光度(ab-sorbance,A)或透射比(transmittance,T)为纵坐标所绘的曲线。
(nm)为横坐标、以吸光度(ab-sorbance,A)或透射比(transmittance,T)为纵坐标所绘的曲线。
2.吸收峰
吸收峰(absorption
peak)是吸收曲线上吸光度最大的地方,它所对应的波长称为最大吸收波长(![]() )。
)。
3.谷
谷(valley)是峰与峰之间的最低部位,其对应的波长称最小吸收波长(![]() )。
)。
4.肩峰
在一个峰旁边产生的曲折,称为肩峰(shoulder peak)。
5.未端吸收
在谱图短波端呈现强吸收但不成峰形的部分,称为末端吸收(end absorption)。
6.生色团
生色团(chromophore)是指有机化合物分子中含有能产生![]() →
→![]() 或
或![]() →
→![]() 跃迁的、能在紫外可见光范围内产生吸收的基团,如C=C、C=O、—C=S、—NO2、—N=N—等。
跃迁的、能在紫外可见光范围内产生吸收的基团,如C=C、C=O、—C=S、—NO2、—N=N—等。
7.助色团
助色团(auxochrome)是含有非键电子对的杂原子饱和基团,当它们与生色团或饱和烃相连时,能使生色团或饱和烃的吸收峰向长波方向移动,并使吸收强度增加,如—OH、—NH2、—SH、—X(卤素)、—OR等。
8.红移
红移(red shift)是指由于化合物的结构改变,如引入助色团、发生其轭作用以及改变溶剂等,使吸收峰向长波方向移动。
9.蓝(紫)移
蓝移(blue shift)是指当化合物的结构改变或受溶剂影响,使吸收峰向短波方向移动。
10.增色效应和减色效应
由于化合物结构改变或其他原因,使吸收强度增强,称增色效应(hyperchromic effect);使吸收强度减弱,称减色效应(hypochromic effect)。
紫外-可见光谱吸收带的位置易受分子中结构因素和测定条件等多种因素的影响,其核心是对分子中电子共轭结构的影响。
1.共轭效应
共轭体系的形成使分子的最高己占轨道能级升高,最低空轨道能级降低,![]() →
→![]() 跃迁的能量降低。共轭体系越长,
跃迁的能量降低。共轭体系越长,![]() 和
和![]() 轨道的能量差越小,最大吸收波长越移向长波方向,吸收强度也增大。
轨道的能量差越小,最大吸收波长越移向长波方向,吸收强度也增大。
2.立体化学效应
立体化学效应(stereochemical effect)是指因空间位阻、构象、跨环共轭等因素导致吸收光谱的红移或蓝移,并常伴随有增色或减色效应。
空间位阻(steric hindrance)妨碍分子内共轭的发色基团处于同一平面,使共轭效应(con-jugative effect)减小甚至消失,从而影响吸收带波长的位置。
跨环效应(cross-ring
effect)是指两个发色基团虽不共轭,但由于空间的排列,使它们的电子云仍能相互影响,使![]() 和
和![]() 改变。
改变。
3.溶剂的影响
在溶液中溶质分子是溶剂化的,限制了溶质分子的自由转动,使转动光谱消失。溶剂的极性增大,使溶质分子的振动受限制,由振动引起的光谱精细结构亦消失。当物质溶解在非极性溶剂中时,其光谱与物质气态的光谱较相似,可以呈现孤立分子产生的转动-振动精细结构。
溶剂极性的不同也会引起某些化合物吸收光谱的红移或蓝移,这种作用称为溶剂效应(solvent
effect)。在![]() →
→![]() 跃迁中,激发态极性大于基态,当使用极性大的溶剂时,由于溶剂导致吸收谱带
跃迁中,激发态极性大于基态,当使用极性大的溶剂时,由于溶剂导致吸收谱带![]() 红移。而在
红移。而在![]() →
→![]() 跃迁中,基态n电子与极性溶剂形成氢键,降低了基态能量,使激发态与基态之间的能量差变大,导致吸收带
跃迁中,基态n电子与极性溶剂形成氢键,降低了基态能量,使激发态与基态之间的能量差变大,导致吸收带![]() 向短波区移动(蓝移)。
向短波区移动(蓝移)。
图3.5给出了在极性溶剂中![]() →
→![]() 和
和![]() →
→![]() 跃迁能量变化示意图。
跃迁能量变化示意图。
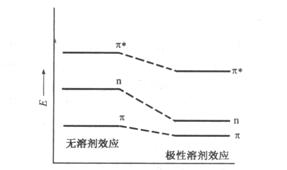
图3.5 溶剂极性对![]() →
→![]() 和
和![]() →
→![]() 跃迁能量的影响
跃迁能量的影响
4.体系pH的影响
无论是酸性、碱性或中性介质,体系的pH对紫外-可见光谱的影响是普遍的现象。如酚类化合物由于体系的pH不同,其解离情况不同,紫外光谱也不同
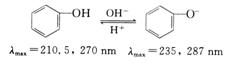
有关体系pH对显色反应的影响,可参阅
3.2 吸收光谱的测量—朗伯-比尔定律
当一束平行光通过均匀的液体介质时,光的一部分被吸收,一部分透过溶液,还有一部分被器皿表面反射。设入射光强度为![]() ,吸收光强度为
,吸收光强度为![]() a透射光强度为
a透射光强度为![]() ,反射光强度为
,反射光强度为![]() ,则
,则
![]() (3.3)
(3.3)
在吸收光谱分析中,被则溶液和参比溶液一般是分别放在同样材料和厚度的吸收池中,让强度为![]() 的单色分别通过个吸收池,再测量透射光的强度。所以反射光的影响可相互抵消,式(3.3)可简化为
的单色分别通过个吸收池,再测量透射光的强度。所以反射光的影响可相互抵消,式(3.3)可简化为
![]() (3.4)
(3.4)
透射光的强度![]() 与入射光强度(
与入射光强度(![]() )之比称透射比,或称透光度,用
)之比称透射比,或称透光度,用![]() 表示,则有
表示,则有
![]() (3.5)
(3.5)
溶液的透射比越大,表示它对光的吸收越小;反之,透射比越小,表示它对光的吸收越大。常用吸光度来表示物质对光的吸收程度,其定义为
![]() (3.6)
(3.6)
![]() 值越大,表明物质对光的吸收越大。透射比和吸光度都是表示物质对光的吸收程度的一种量度,透射比常以百分率表示,称为百分透射比,
值越大,表明物质对光的吸收越大。透射比和吸光度都是表示物质对光的吸收程度的一种量度,透射比常以百分率表示,称为百分透射比,![]() ;吸光度是一个量纲为一的量,两者可由式(3.6)相互换算。
;吸光度是一个量纲为一的量,两者可由式(3.6)相互换算。
朗伯-比尔(Lambert-Beer)定律是光吸收的基本定律,也是分光光度分析法的依据和基础。当入射光波长一定时,溶液的吸光度![]() 是待测物质浓度和液层厚度的函数。Lambert和Beer分别于1760年和1852年研究了溶液的吸光度与溶液层厚度和溶液浓度之间的定量关系。当用适当波长的单色光照射一固定浓度的溶液时,其吸光度与光透过的液层厚度呈正比,此即Lambert定律,其数学表达式为
是待测物质浓度和液层厚度的函数。Lambert和Beer分别于1760年和1852年研究了溶液的吸光度与溶液层厚度和溶液浓度之间的定量关系。当用适当波长的单色光照射一固定浓度的溶液时,其吸光度与光透过的液层厚度呈正比,此即Lambert定律,其数学表达式为
![]() (3.7)
(3.7)
式中:![]() 为比例系数,
为比例系数,![]() 为液层厚度(即样品的光程长度)。Lambert定律适用于任何非散射的均匀介质,但它不能阐明吸光度与溶液浓度的关系。
为液层厚度(即样品的光程长度)。Lambert定律适用于任何非散射的均匀介质,但它不能阐明吸光度与溶液浓度的关系。
Beer定律描述了溶液浓度与吸光度之间的定量关系。当用一适当波长的单色光照射厚度一定的均匀溶液时,吸光度与溶液浓度呈正比,即
![]() (3.8)
(3.8)
式中:![]() 为溶液浓度,
为溶液浓度,![]() 为比例系数。
为比例系数。
当溶液的浓度(![]() )和液层的厚度(
)和液层的厚度(![]() )均可变时,它们都会影响吸光度的数值。合并(3.7)和(3.8)两式,得到Lambert-Beer定律,其数学表达式为
)均可变时,它们都会影响吸光度的数值。合并(3.7)和(3.8)两式,得到Lambert-Beer定律,其数学表达式为
![]() (3.9)
(3.9)
在式(3.9)中的比例系数![]() 的值及单位与
的值及单位与![]() 和
和![]() 采用的单位有关。
采用的单位有关。![]() 的单位通常以cm表示,因此
的单位通常以cm表示,因此![]() 的单位主要决定于浓度
的单位主要决定于浓度![]() 用什么单位。
用什么单位。![]() 以g·L-1为单位时,
以g·L-1为单位时,![]() 称为吸光系数(ab-sorptivity),以
称为吸光系数(ab-sorptivity),以![]() 表示,单位为L·g-1·cm-1。当
表示,单位为L·g-1·cm-1。当![]() 以mol·L-1为单位时,
以mol·L-1为单位时,![]() 称为摩尔吸光系数(molar
absorptivity),符号
称为摩尔吸光系数(molar
absorptivity),符号![]() ,单位为L·mol-1·cm-1。当吸收介质内只有一种吸光物质时,式(3.9)表赤这
,单位为L·mol-1·cm-1。当吸收介质内只有一种吸光物质时,式(3.9)表赤这
![]() (3.10)
(3.10)
![]() 比
比![]() 更常用,因为有时吸收光谱的纵坐标用
更常用,因为有时吸收光谱的纵坐标用![]() 或1g
或1g![]() 表示,并以最大摩尔吸光系数(
表示,并以最大摩尔吸光系数(![]() )表示吸光强度。摩尔吸光系数在特定波长和溶剂的情况下是吸光质点的一个特征参数,在数值上等于吸光物质的浓度为1mol·L-1、液层厚度为
)表示吸光强度。摩尔吸光系数在特定波长和溶剂的情况下是吸光质点的一个特征参数,在数值上等于吸光物质的浓度为1mol·L-1、液层厚度为![]() 越大,方法的灵敏度越高。如
越大,方法的灵敏度越高。如![]() 为104数量级时,测定该物质的浓度范围可以达到10-6~10-5mol·L-1,当
为104数量级时,测定该物质的浓度范围可以达到10-6~10-5mol·L-1,当![]() <103时,其测定范围在10-4~10-3mol·L-1左右。
<103时,其测定范围在10-4~10-3mol·L-1左右。
![]() 一般是由较稀浓度溶液的吸光度计算求得,由于
一般是由较稀浓度溶液的吸光度计算求得,由于![]() 与入射光波长有关,因此在表示某物质溶液的
与入射光波长有关,因此在表示某物质溶液的![]() 时,常用下标注明入射光的波长。
时,常用下标注明入射光的波长。
在化合物的组成成分不明的情况下,物质的摩尔质量也不知道,因而物质的量浓度无法确定,就无法使用摩尔吸光系数。在这种情况下![]() 用
用![]() ·(100mL)-1表示,可采用比吸光系数(specific
absorptivity)这一概念。比吸光系数是指物质的质量分数为1%,
·(100mL)-1表示,可采用比吸光系数(specific
absorptivity)这一概念。比吸光系数是指物质的质量分数为1%,![]() 为
为![]() 表示。
表示。![]() 与
与![]() 和
和![]() 的关系为
的关系为
![]() =10
=10![]() =
=![]() (3.11)
(3.11)
式中:![]() 为吸光物质的摩尔质量。
为吸光物质的摩尔质量。
如果溶液中同时存在两种或两种以上吸光物质时,只要共存物质不互相影响性质,溶液的吸光度将是各组分吸光度的总和
![]() (3.12)
(3.12)
在一均匀体系中,当物质浓度固定时,吸光度![]() 与样品的光程长
与样品的光程长![]() 之间的线性关系(Lambert定律)总是普遍成立而无一例外。但在
之间的线性关系(Lambert定律)总是普遍成立而无一例外。但在![]() 恒定时,吸光度
恒定时,吸光度![]() 与浓度
与浓度![]() 之间正比关系有时可能失效,也就是说会偏离Lambert-Beer定律。一般以负偏离的情况居多,因而影响了测定的准确度。引起偏离Lambert-Beer定律的因素很多,通常可归成两类,一类与样品有关,另一类则与仪器有关,分别叙述如下。
之间正比关系有时可能失效,也就是说会偏离Lambert-Beer定律。一般以负偏离的情况居多,因而影响了测定的准确度。引起偏离Lambert-Beer定律的因素很多,通常可归成两类,一类与样品有关,另一类则与仪器有关,分别叙述如下。
1.与测定样品溶液有关的因素
通常只有当溶液浓度小于0.01mol·L-1的稀熔液中Lambert-Beer定律才能成立。在高浓度时,由于吸光质点间的平均距离缩小,邻近质点彼此的电荷分布会产生相互影响,以致改变它们对特定辐射的吸收能力,即吸光系数发生改变,导致对Beer定律的偏离。
推导Lambert-Beer定律时隐含着测定试液中各组分之间没有相互作用的假设。但随着溶液浓度增加,各组分之间的相互作用则是不可避免的。例如,可以发生离解、缔合、光化反应、互变异构及络合物配位数变化等作用,会使被测组分的吸收曲线发生明显改变,吸收峰的位置、高度以及光谱精细结构等都会不同,从而破坏了原来的吸光度与浓度的函数关系,偏离了Beer定律。
溶剂对吸收光谱的影响也很重要。在分光光度法中广泛使用各种溶剂,它会对生色团的吸收峰高度、波长位置产生影响。溶剂还会影响待测物质的物理性质和组成,从而影响其光谱特性,包括谱带的电子跃进类型等。
当试样为胶体、乳状液或有悬浮物质存在时,入射光通过溶液后,有一部分光会因散射而损失,使吸光度增大,对Beer定律产生正偏差。质点的散射强度是与入射光波长的4次方呈反比的,所以散射对紫外区的测定影响更大。
2.与仪器有关的因素
严格讲Lambert-Beer定律只适用于单色光,但在紫外-可见分光光度法中从光源发出的光经单色器分光,为满足实际测定中需有足够光强的要求,狭缝必须有一定的宽度。因此,由出射狭缝投射到被测溶液的光,并不是理论上要求的单色光。这种非单色光是所有偏离Beer定律的因素中较为重要的因素之一。因为实际用于测量的是一小段波长范围的复合光,由于吸光物质对不同波长的光的吸收能力不同,就导致了对Beer定律的负偏离。在所使用的波长范围内,吸光物质的吸光系数变化越大,这种偏离就越显著。例如,按图3.6所示的吸收光谱,谱带I的吸光系数变化不大,用谱带I进行分析,造成的偏离就比较小。而谱带II的吸光系数变化较大,用谱带II进行分析就会造成较大的负偏离。所以通常选择吸光物质的最大吸收波长作为分析波长。这样不仅能保证测定有较高的灵敏度,而且此处曲线较为平坦,吸光系数变化不大,对Beer定律的偏离程度就比较小。并且在保证一定光强的前提下,应使用尽可能窄的有效带宽宽度,同时应尽量避免采用尖锐的吸收峰进行定量分析。
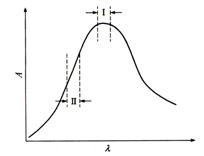
图3.6 分析谱带的选择
3.3 紫外-可见分光光度计
各种型号的紫外-可见分光光度计(UV-Vis spectrophotometer),就其结构来说,都是由五部分组成(见图3.7),即光源(light source)、单色器(monochromator)、吸收池(absorptioncell)、检测器(detector)和信号指示系统(signal indicating system)。
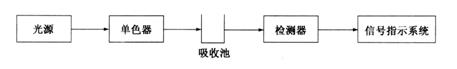
图3.7 紫外-可见分光光度计基本结构示意图
紫外-可见分光光度计可归纳为5种类型,即单光束分光光度计、双光束分光光度计、双波长分光光度计、多通道分光光度计和探头式分光光度计。前三种类型较为普遍。
1.单光束分光光度计
单光束分光光度计(single beam spectrophotometer)的光路示意图3.7,经单色器分光后的一束平行光,轮流通过参比溶液和样品溶液,以进行吸光度的测定。这种简易型分光光度计结构简单,操作方便,维修容易,适用于常规分析。
2.双光束分光光度计
双光束分光光度计(double beam spectrophotometer)的光路示意于图3.8。经单色器分光后经反射镜(M1)分解为强度相等的两束光,一束通过参比池,另一束通过样品池。双光束分光光度计能自动比较两束光的强度,此比值即为试样的透射比,经对数变换将它转换成吸光度并作为波长的函数记录下来。由于两束光同时分别通过参比池和样品池,还能自动消除光源强度变化所引起的误差。
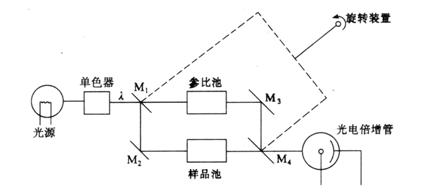
图3.8 单波长双光束分光光度计原理图
M1 ,M2, M3
,M4.反射镜
3.双波长分光光度计
双波长分光光度计(double
wavelength spectrophotometer)基本光路如图3.9所示。由同一光源发出的光被分成两束,分别经过两个单色器,得到两束不同波长(![]() 和
和![]() )的单色光;利用切光器使两束光以一定的频率交替照射同一吸收池,然后经过光电倍增管和电子控制系统,最后由显示器显示出两个波长处的吸光度差值
)的单色光;利用切光器使两束光以一定的频率交替照射同一吸收池,然后经过光电倍增管和电子控制系统,最后由显示器显示出两个波长处的吸光度差值![]() 。
。
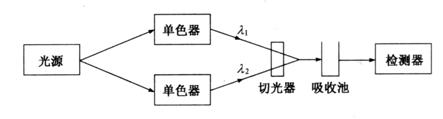
图3.9 双波长分光光度计光路示意图
对于多组分混合物、混浊试样(如生物组织液)分析,以及存在背景干扰或共存组分吸干扰的情况下,利用双波长分光光度法,往往能提高方法的灵敏度和选择性。利用双波长分光光度计,能获得导数光谱。通过光学系统转换,使双波长分光光度计能很方便地转化为单波长工作方式。如果能在![]() 和
和![]() 处分别记录吸光度随时间变化的曲线,还能进行化学反应动力学研究。
处分别记录吸光度随时间变化的曲线,还能进行化学反应动力学研究。
4.多通道分光光度计
多通道分光光度计(multichannel spectrophotometer)的光路原理如图3.10所示。由于光源发射出的复合光先通过样品池后再经全息光栅色散,色散后的单色光由光二极管阵列中的光二极管接收,能同时检测190~900nm波长范围,因此在极短的时间内(≤1s)给出整个光谱的全部信息。这种光度计特别适于进行快速反应动力学研究和多组分混合物的分析,也己被用作高效液相色谱和毛细管电泳仪的检测器。
5.光导纤维探头式分光光度计
图3.11是光导纤维探头式分光光度计(optical
fiber probe-type spectrophotometer)的光路示意图。探头由两根相互隔离的光导纤维组成。由钨灯发射的光由其中一根光纤传导致试样溶液,再经反射镜反射后由另一根光纤传导,通过干涉滤光片后由光电二级管接收转变为电信号。这类光度计不需要吸收池,直接将探头插入样品溶液中进行原位检测,不受外界光线的影响。这类光度计常用于环境和过程分析。
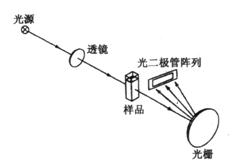
图3.10 光学多通道分光光度计光路示意图
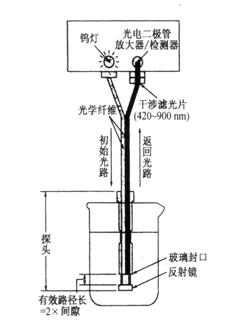
图3.11 光导纤维探头式分光光度计光路图
通常在实验室工作中,验收新仪器或仪器使用过一段时间后都要进行波长校正和吸光度校正。建议采用下述的较为简便和实用的方法来进行校正。
镨钕玻璃或钬玻璃都有若干特征的吸收峰,可用来校正分光光度计的波长标尺。前者用于可见光区,后者则对紫外和可见光区都适用。
可用K2CrO4标准溶液来校正吸光度标度。将
表3.2 铭酸钾溶液的吸光度
|
|
吸光度 |
|
吸光度 |
|
吸光度 |
|
吸光度 |
|
220 |
0.4559 |
300 |
0.1518 |
380 |
0.9281 |
460 |
0.0173 |
|
230 |
0.1675 |
310 |
0.0458 |
390 |
0.6841 |
470 |
0.0083 |
|
240 |
0.2933 |
320 |
0.0620 |
400 |
0.3872 |
480 |
0.0035 |
|
250 |
0.4962 |
330 |
0.1457 |
410 |
0.1972 |
490 |
0.0009 |
|
260 |
0.6345 |
340 |
0.3143 |
420 |
0.1261 |
500 |
0.0000 |
|
270 |
0.7447 |
350 |
0.5528 |
430 |
0.0841 |
|
|
|
280 |
0.7235 |
360 |
0.8297 |
440 |
0.0535 |
|
|
|
290 |
0.4295 |
370 |
0.9914 |
450 |
0.0325 |
|
|
3.4 分析条件的选择
为使用分析方法有较高的灵敏度和准确度,选择最佳的测定条件是很重要的。这些条件包括仪器测量条件、试样反应条件以及参比溶液的选择等。
任何光度计都有一定的测量误差,这是由于光源不稳定、实验条件的偶然变动、读数不准确等因素造成的。这些因素对于试样的测定结果影响较大,特别是当试样浓度大或较小时。因此要选择适宜的吸光度范围,以使测量结果的误差尽量减小。根据Lambert-Beer定律
![]()
微分手,得
![]()
或
![]() (3.13)
(3.13)
将式(3.13)代入Lambert-Beer定律,则测定结果的相对误差为
![]() (3.14)
(3.14)
要使测定结果的相对误差(![]() )最小,对
)最小,对![]() 求导数应有一极小值,即
求导数应有一极小值,即
![]() (3.15)
(3.15)
解得
![]() 或
或 ![]()
即当吸光度![]() 时,吸光度测量误差最小。上述结果也可由图3.12表示,即图中曲线的最低点。如果光度计读数误差为1%,若要求浓度测量的相对误差小于5%,则待测溶液的透射比应选在70%~10%范围内,吸光度为0.15~1.00。实际工作中,可通过调节待测溶液的浓度,选用适当厚度的吸收池等方式使透射比
时,吸光度测量误差最小。上述结果也可由图3.12表示,即图中曲线的最低点。如果光度计读数误差为1%,若要求浓度测量的相对误差小于5%,则待测溶液的透射比应选在70%~10%范围内,吸光度为0.15~1.00。实际工作中,可通过调节待测溶液的浓度,选用适当厚度的吸收池等方式使透射比![]() (或吸光度
(或吸光度![]() )落在此区间内。现在高档的分光光度计使用性能优越的检测器,即使吸光度高达2.0,甚至3.0时,也能保证浓度测量的相对误差小于5%。
)落在此区间内。现在高档的分光光度计使用性能优越的检测器,即使吸光度高达2.0,甚至3.0时,也能保证浓度测量的相对误差小于5%。
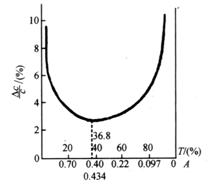
图3.12 浓度测量的相对误差
![]() 与溶液透射比(T)或吸光度(A)的关系
与溶液透射比(T)或吸光度(A)的关系
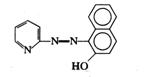
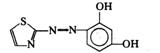
在无机分析中,很少利用金属离子本身的颜色进行光度分析,因为它们的吸光系数值都比较小。一般都是选用适当的试剂,与待测离子反应生成对紫外可见光有较大吸收的物质再行测定。这种反应称为显色反应,所用的试剂称为显色剂。络合反应、氧化还原反应以及增加生色基团的衍生化反应等都是常见的显色反应类型,尤以络合反应应用最广。许多有机显色剂与金属离子形成稳定性好、具有特征颜色的螯合物,其灵敏度和选择性都较高。表3.3列举了几种显色剂及其有色配合物。
表3.3 一些常用的显色剂
|
|
试剂 |
结构式 |
离解常数 |
测定离子 |
|
无机显色剂 |
硫氰酸盐 钼酸盐 过氧化氢 |
SCN-
H2O2 |
pKa=0.85 pKa2=3.75 pKa=11.75 |
Fe2+,Mo(V),W(V) Si(IV),P(V) Ti(IV) |
|
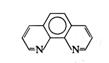
有机显色剂 |
邻二氮菲 |
|
pKa=4.96 |
Fe2+ |
|
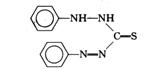
双硫踪 |
|
pKa=4.6 |
Pb2+,Hg2+,Zn2+,Bi3+等 |
|
|
丁二酮肟 |
|
pKa=10.54 |
Ni2+,Pd2+ |
|
|
铬天青S (CAS) |
|
pKa3=2.3 pKa4=4.9 pKa5=11.5 |
Be2+,A13+,Y3+, Ti4+Zr4+,Hf4+ |
|
|
茜素红S |
|
pKa2=5.5 pKa3=11.0 |
Al3+,Ga3+,Zr(IV), Th(IV),F-,Ti(IV) |
|
|
偶氮胂III* |
|
|
UO Zr(IV),RE3+,Y3+, Sc3+,Ca2+等 |
|
|
4-(2-吡啶偶氮)-间苯二酚(PAN) |
|
pKa1=3.1 pKa2=5.6 pKa3=11.9 |
Co2+,Pb2+, Ga3+,Nb(V),Ni2+ |
|
|
1-(2-吡喧偶氮)-2-萘酚(PAN) |
|
pKa1=2.9 pKa2=11.2 |
Co2+,Ni2+,Zn2+,Pb2+ |
|
|
4-(2-噻唑偶氮)-间苯二酚 (TAR) |
|
|
Co2+,Ni2+,Cu2+,Pb2+ |
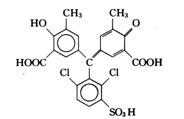
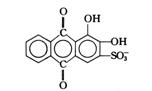
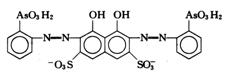
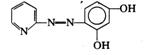
![]()
![]()
显色反应一般应满足下述要求:
(1)反应的生成物必须在紫外、可见光区有较强的吸光能力,即摩尔吸光系数较大,反应有较高的选择性;
(2)反应生成物应当组成恒定、稳定性好,显色条件易于控制等,这样才能保证测量结果有良好的重要性;
(3)对照性要好,显色剂与有色配合物的![]() 的差别要在60nm以上。
的差别要在60nm以上。
实际上能同时满足上述条件的显色反应不很多,因此在初步选定好显色剂以后,认真细致地研究显色反应的条件十分重要。下面介绍其主要影响因素。
1.显色剂用量
生成络合物的显色反应可用下式表示
M
+ nR =MRn
![]() (3.16)
(3.16)
式中:M代表金属离子,R为显色剂,βn为配合物的累积稳定常数。由式(3.16)可见,当[R]固定时,从M转化成MR![]() 的转化率将不发生变化。对稳定性好的(即βn大)配合物,只要显色剂过量,显色反应即能定量进行。而对不稳定的配合物或可形成逐级配合物时,显色剂用量要过量很多或必须严格控制。例如,以SCN
的转化率将不发生变化。对稳定性好的(即βn大)配合物,只要显色剂过量,显色反应即能定量进行。而对不稳定的配合物或可形成逐级配合物时,显色剂用量要过量很多或必须严格控制。例如,以SCN![]() 作显色剂测定钼时,要求生成红色的Mo(SCN)5配合物进行测定。但当SCN
作显色剂测定钼时,要求生成红色的Mo(SCN)5配合物进行测定。但当SCN![]() 浓度过高时,由于会生成浅红色的Mo(SCN)
浓度过高时,由于会生成浅红色的Mo(SCN)![]() 配合物而使吸光度降低。又如,用铁的硫氰酸配合物测定Fe(III)时,随SCN
配合物而使吸光度降低。又如,用铁的硫氰酸配合物测定Fe(III)时,随SCN![]() 浓度增大,逐步形成颜色更深的不同配位数的化合物,吸光度增加。因此在这两种离子的测定中必须严格控制显色剂用量,才能得到准确的结果。显色剂的用量可通过实验确定,作吸光度随显色剂浓度变化曲线,选恒定吸光度值时的显色剂用量。
浓度增大,逐步形成颜色更深的不同配位数的化合物,吸光度增加。因此在这两种离子的测定中必须严格控制显色剂用量,才能得到准确的结果。显色剂的用量可通过实验确定,作吸光度随显色剂浓度变化曲线,选恒定吸光度值时的显色剂用量。
2、溶液酸度的影响
多数显色剂都是有机弱酸或弱碱,介质的酸度会直接影响显色剂的离解程度,从而影响显色反应的完全程度。溶液酸度的影响表现在许多方面。
(1)由于pH不同,可形成具有不同配位数,不同颜色的化合物。金属离子与弱酸阴离子在酸性溶液中大多生成低配位数的络合物,可能并没有达到阳离子的最大配位数。当pH增大时,游离的阴离子浓度相应增大,使得可能生成高配位数的化合物。例如,Fe(III)可与水杨酸在不同pH生成组成配比不同的配合物(见下表):
|
pH范围 |
配合物组成 |
颜色 |
|
<4 |
Fe(C7H4O3) |
紫红色(1:1) |
|
4~7 |
Fe(C7H4O3) |
棕橙色(1:2) |
|
8~10 |
Fe(C7H4O3)33- |
黄 色(1:3) |
在用这类反应进行测定时,控制溶液的pH至关重要。
(2)pH增大会引起某些金属离子水解而形成各种型体的羟基配合物,甚至可能析出沉淀;或者由于生成金属的氢氧化物而破坏了有色配合物,使溶液的颜色完全退去,例如
![]()
![]() Fe(SCN)2+ +
OH-
Fe(SCN)2+ + OH-
Fe(SCN)2+ +
OH-
Fe(SCN)2+ + OH-
显色反应的最宜酸度可估算如下,如果金属离子与配位体R生成逐级配合物MR![]() ,即
,即
![]()
![]() M + nR
MRn
M + nR
MRn
条件累积稳定常数![]() 和累积稳定常数
和累积稳定常数![]() 有如下关系式
有如下关系式
![]()
![]() (3.17)
(3.17)
式中:![]() 和
和![]() 分别为M和R的副反应系数。当上述反应定量进行时(即99.9%的M转化为MR
分别为M和R的副反应系数。当上述反应定量进行时(即99.9%的M转化为MR![]() ),则
),则
![]() (3.18)
(3.18)
即要求![]() 。
。
以邻二氮菲(Phen)与Fe(II)的显色反应为例。假定反应在0.1mol·L-1柠檬酸盐![]() 缓冲溶液中进行,过量显色剂浓度[Phen
缓冲溶液中进行,过量显色剂浓度[Phen![]() ]为10-4mol·L-1,Fe-Phen络合物的1g
]为10-4mol·L-1,Fe-Phen络合物的1g![]() 为 21.3,不同pH时的1g
为 21.3,不同pH时的1g![]() [Fe(A)]和1g
[Fe(A)]和1g![]() [Phen(H)]见表3.4。
[Phen(H)]见表3.4。
表3.4 酸度对显色反应完全度的影响
|
pH |
|
|
|
|
|
1 |
— |
3.9 |
9.6 |
—2.4 |
|
2 |
— |
2.9 |
12.6 |
0.6 |
|
3 |
— |
1.9 |
15.6 |
3.6 |
|
4 |
0.5 |
1.0 |
17.8 |
5.8 |
|
5 |
2.6 |
0.3 |
17.8 |
5.8 |
|
6 |
4.2 |
— |
17.1 |
5.1 |
|
7 |
5.5 |
— |
15.8 |
3.8 |
|
8 |
6.5 |
— |
14.8 |
2.8 |
|
9 |
7.5 |
— |
13.8 |
1.8 |
|
10 |
8.5 |
— |
12.8 |
0.8 |
各pH下的![]() 可按下式计算
可按下式计算
![]()
![]()
其计算结果(见表3.4)表明,在柠檬酸盐缓冲溶液中邻二氮菲与Fe(II)显色反应的最宜pH范围是3~8,这与实验结果基本一致。
实际工作中是通过实验来确定显色反应的最宜酸度的。具体做法是固定溶液中待测组分与显色剂的浓度,改变溶液的酸度(pH),测定溶液的吸光度![]() 与pH的关系曲线,从中找出最宜pH范围。
与pH的关系曲线,从中找出最宜pH范围。
3.其他问题
显色反应的时间、温度、放置时间对络合物稳定性的影响等都对显色反应有影响。这些都需要通过条件试验来确定。
测量试样溶液的吸光度时,先要用参比溶液调节透射比为100%,以消除溶液中其他成分以及吸收池和溶剂对光的反射和吸收所带来的误差。根据试样溶液的性质,选择合适组分的参比溶液是很重要的。
1.溶剂参比
当试样溶液的组成较为简单,共存的其他组分很少且对测定波长的光几乎没有吸收时,可采用溶剂作为参比溶液,这样可消除溶剂、吸收池等因素的影响。
2.试剂参比
如果显色剂或其他试剂在测定波长有吸收,按显色反应相同的条件,只是不加入试样,同样加入试剂和溶剂作为参比溶液。这种参比溶液可消除试剂中的组分产生吸收的影响。
3.试样参比
如果试样基体在测定波长有吸收,而与显色剂不起显色反应时,可按与显色反应相同的条件处理试样,只是不加显色剂。这种参比溶液适用于试样中有较多的共存组分,加入的显色剂量不大,且显色剂在测定波长无吸收的情况。
4.平行操作溶液参比
用不含被测组分的试样,在相同条件下与被测试样同样进行处理,由此得到平行操作参比溶液。
在光度分析中,体系内存在的干扰物质的影响有以下几种情况:①干扰物质本身有颜色或显色剂形成有色化合物,在测定条件下也有吸收;②在显色条件下,干扰物质水解,析出沉淀使溶液混浊,致使吸光度的测定无法进行;③与待测离子或显色剂形成更稳定的配合物,使显色反应不能进行完全。
可以采用以下几种方法来消除这些干扰作用:
(1)控制酸度 根据配合物的稳定性不同,可以利用控制酸度的方法提高反应的选择性,以保证主反应进行完全。例如,双硫腙能与Hg2+、Pb2+、Cu2+、Ni2+、Cd2+等十多种金属离子形成有色配合物,其中与Hg2+生成的络合物最稳定,在0.5mol·L-1H2SO4介质中仍能定量进行,而上述其他离子在此条件下不发生反应。
(2)选择适当的掩蔽剂 使用掩蔽剂消除干扰是常用的有效方法。选取的条件是掩蔽剂不与待测离子作用,掩蔽剂以及它与干扰物质形成的配合物的颜色应不干扰待测离子的测定。
(3)利用生成惰性络合物 例如钢铁中微量钴的测定,常用钴试剂为显色剂。但钴试剂不仅与Co2+有灵敏的反应,而且与Ni2+、Zn2+、Mn2+、Fe2+等都有反应。但它与Co2+在弱酸性介质中一旦完成反应后,即使再用强酸酸化溶液,该络合物也不会分解。而Ni2+、Zn2+、Mn2+、Fe2+等与钴试剂形成的络合物在强酸介质中很快分解,从而消除了上述离子的干扰,提高子反应的选择性。
(4)选择适当的测量波长 如在K2Cr2O7存在下测定KMnO4时,不是选![]() (525nm),而是选
(525nm),而是选![]() =545 nm。这样测定KMnO4溶液的吸光度,K2Cr2O7就不干扰了。
=545 nm。这样测定KMnO4溶液的吸光度,K2Cr2O7就不干扰了。
(5)分离 若上述方法不宜采用时,也可以采用预先分离的方法,如沉淀、萃取、离子交换、蒸发和蒸馏以及色谱分离法(包括柱色谱、绝色谱、薄层色谱等)。
此外,还可以利用化学计量学方法实现多组分同时测定,以及利用导数光谱法,双波长光谱法等新技术来消除干扰。
3.5 紫外-可见分光光度法的应用
紫外-可见分光光度法是对物质进行定性分析、结构分析和定量分析的一种手段,而且还能测定某些化合物的物理化学参数,例如摩尔质量、络合物的络合比和稳定常数,以及酸、碱离解常数等。
紫外-可见分光光度法较少用于无机元素的定性分析,无机元素的定性分析可用原子发射光谱法或化学分析的方法。在有机化合物的定性鉴定和结构分析中,由于紫外-可见光谱较简单,特征性不强,因此该法的应用也有一定的局限性。但是它适用于不饱和有机化合物,尤其是其轭体系的鉴定,以此推断未知物的骨架结构。此外,可配合红外光谱、核磁共振波谱法和质谱法进行定性鉴定和结构分析,因此它仍不失为是一种有用的辅助方法。
目前,己有多种以实验结果为基础的各种有机化合物的紫外-可见光谱标准谱图,有的则汇编了有关电子光谱的数据表。常用的标准谱图有以下几种:
(1)Sadtler Standard spectra(Ultraviolet).London: Heyden,1978
(2)Frieded R A and Orchin M. Ultraviolet Spectra Of Aromatic Compounds.New York: Wiley,1951
(3)Kenzo Hirayama。Handbook of Ultraviolet and Visible Absorption Spectra of Organic Compounds. New York: Plenum, 1967
*(4)Organic Electronic Spectral Data. John Wiley and Sons, 1946~
*这是一套由许多作者共同编写的大型手册性丛书,所搜集的文献资料自1946年开始,目前还在继续编写。
应该指出,分子或离子对紫外-可见光的吸收只是它们含有生色基团和助色基团的特征,而不是整个分子或离子的特征。因此仅靠紫外-可见光谱来确定一个未知物的结构是不现实的,还要参照Woodward-Fieser规则和Scott规则以及其他方法的配合。Woodward-Fieser规则和Scott规则都是经验规则,当用其他的物理和化学方法判断某化合物的几种可能结构时,可用它们来计算最大吸收波长![]() ,并与实验值进行比较,以确认物质的结构。
,并与实验值进行比较,以确认物质的结构。
1.Woodward-Fieser规则
Woodward提出了计算共轭二烯、多烯烃及共轭烯酮类化合物![]() →
→![]() ﹡迁最大吸收波长的经验规则,如表3.5和表3.6所示。计算时先从母体得到一个最大吸收的基数,然后对连接在母体
﹡迁最大吸收波长的经验规则,如表3.5和表3.6所示。计算时先从母体得到一个最大吸收的基数,然后对连接在母体![]() 电子体系上的不同取代基以及其他结构因素加以修正。
电子体系上的不同取代基以及其他结构因素加以修正。
表3.5 计算二烯烃或多烯烃 的最大吸收位置(己烷为溶剂)
|
化合物 |
|
|
母体是异环的二烯烃或无环多烯烃类型
母体是同环的二烯烃或这种类的多烯烃*
增加1个共轭双键 环外双键 每个烷基取代基 每个极性基 —O—乙酰基 —O—R —S—R —Cl,—Br —NR2 溶剂校正值 |
基值214 基值253 30 5 5 0 6 30 5 60 0 |
*当两种情形的二烯烃体系同时存在时,选择波长较长的为其母体系统,即选用基值为253nm
(1)几种化合物的![]() 计算汇列
计算汇列
![]() [例3.1]
[例3.1]
解: 基值
253nm
环外双键
5nm
烷基取代基(3×5)
15nm
273nm
![]() [例3.2]
[例3.2]
解: 基值
253nm
烷基取代基(4×5) 20nm
环外双键
5nm
共轭系统的延长
30nm
308nm
表3.6 计算不饱和羰基化合物![]() 的最大吸收位置
的最大吸收位置
|
|
|
|
醛 当X为HO或RO时 每增加1个共轭双键 同环二烯化合物 环外双键 每个取代烷基
每个极性基
—OH
—OA c
—OR
—SR
—Cl
—Br
—NR2
溶剂校正 乙醇、甲醇 氯仿 二氧六环 乙醚 己烷,环己烷 水 |
215 -13 -6 -22 30 39 5 10 12 18 35 30 50 6 35 30 17 31 85 15 12 25 30 95 0 1 5 7 11 -8 |
[例3.3]
解 基值
253nm
烷基取代基(3×5) 15nm
环外双键
5nm
取代基(OCOCH3)
0
共轭系统的延长
30nm
303nm
![]() [例3.4]
[例3.4]
解 基值
253nm
烷基取代(4×5)
20nm
环外双键(0)
0
共轭系统的延长(1×30) 30nm
303nm
 [例3.5]
[例3.5]
解 基值
214nm
烷基取代(5×5)
25nm
共轭系统的延长(1×30) 30nm
环外双键(2×5)
10anm
279nm
a这个双键是两个环的环外双键,故乘2。
2.Scott规则
Scott规则类似于Woodward规则,用来计算芳香族羰基的衍生物在乙醇溶液中的![]() 表3.7和表3.8列出了该经验规则的计算方法。
表3.7和表3.8列出了该经验规则的计算方法。
表3.7 PhCOR衍生物E2带![]() 的计算
的计算
|
PhCOR生色团母体 |
|
|
R=烷基或环残基(R) =氢(H) =羟基或烷氧基(OH或OR) |
246 250 230 |
表3.8 苯环上邻、间、对位被取代基取代的![]()
|
取代基 |
邻位 |
间位 |
对位 |
|
R(烷基) OH,OR O CI Br NH2 NHAc NR2 |
3 7 11 0 2 13 20 20 |
3 7 20 0 2 13 20 20 |
10 25 78 10 15 58 45 85 |
[例3.6]
解 母体
246nm
间位—OH
7nm
对位—OH
25nm
计算值
278nm
实测值
279nm
[例3.7]
 解 母体
246nm
解 母体
246nm
邻位环残基(a)
3nm
间位—Br
2nm
计算值
251nm
实测值
248nm
可以应用紫外光谱来确定一些化合物的构型和构象。
1.判别顺反异构体
反式异构体空间位阻小,共轭程度较高,其![]() 和
和![]() ,大于顺式异构体。表3.9和表3.10列举了某些有机化合物的顺反异构体的
,大于顺式异构体。表3.9和表3.10列举了某些有机化合物的顺反异构体的![]() 和
和![]() ,其中番茄红素的吸收光谱图见图3.11。
,其中番茄红素的吸收光谱图见图3.11。
表3.9 某些有机化合物的顺反异构体的![]() 和
和![]()
|
化合物 |
顺式 |
反式 |
||
|
|
/ L·mol-1·cm-1 |
|
/ L·mol-1·cm-1 |
|
|
番茄红素 |
440 |
90 000 |
470 |
185 000 |
|
|
|
弱 |
|
|
|
二苯代乙烯 |
280 |
13 500 |
295 |
27 000 |
|
苯代丙烯酸 |
264 |
9 500 |
273 |
20 000 |
|
|
260 |
11 900 |
270 |
20 100 |
|
丁烯二酸二甲酯 |
198 |
26 000 |
214 |
34 000 |
|
偶氮苯 |
295 |
12 600 |
315 |
50 100 |
|
肉桂酸 |
280 |
13 500 |
295 |
27 000 |
|
1-苯基-1,3-丁二烯 |
265 |
14 000 |
280 |
28 300 |
a380nm吸收峰属新番茄红素的顺式乙烯键;470nm为新番茄红素吸收峰,强度较全反式番茄红素弱。
表3.10 某些多环二烯的顺反异构体吸收强度
|
同环双键(顺发) |
异环双键(反式) |
||
|
化合物 |
|
化合物 |
|
|
麦角甾醇 |
11 800 |
麦角甾醇-D |
21 000 |
|
7-脱氢胆甾醇 |
11 400 |
脱氢麦角甾醇 |
19 000 |
|
胆甾-2,4-二烯 |
7 000 |
胆甾-4,6-二烯 |
28 000 |
|
左旋海松酸 |
7 100 |
松香酸 |
16 100 |
2.判别互变异构体
一般共轭体系的![]() 、
、![]() 大于非共轭体系(见表3.11)。例如,乙酰乙酸乙酯有酮式和烯醇式间的互变异构
大于非共轭体系(见表3.11)。例如,乙酰乙酸乙酯有酮式和烯醇式间的互变异构
![]()
在极性溶剂中该化合物以酮式存在,吸收峰弱;而在非极性溶剂正己烷中以烯醇式为主,出现强的吸收峰。
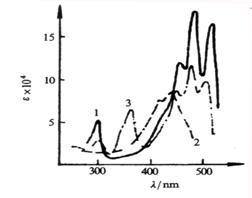
图3.13 蕃茄红素的紫外吸收光谱
![]()
![]()
![]()
![]() 1.全反式蕃茄红素 2 .全反式蕃茄红素 3 .全反式蕃茄红素
1.全反式蕃茄红素 2 .全反式蕃茄红素 3 .全反式蕃茄红素
表3.12 某此有机化合物的互变异构体
|
化合物 |
共轭(醇式) |
非共轭(酮式) |
|
|
|
|
|
亚油酸 |
232 |
无吸收 |
|
苯酰乙酸乙酯 |
308 |
245 |
|
乙酰乙酸乙酯 |
245(18 000) |
240(110) |
|
乙酰丙酮 |
269(12100)(水中) |
277(1900)己烷中 |
|
异丙 |
235(12 000) |
220 |
紫外-可见分光光度法定量分析的依据是Lambert-Beer定律,即在一定波长处被测定物质的吸光度与它的浓度呈线性关系。因此,通过测定溶液对一定波长入射光的吸光度,即可求出该物质在溶液中的浓度或含量。下面介绍几种常用的测定方法。
1.单组分定量方法
(1)校准曲线法 这是实际工作中用得最多的一种方法。具体做法是:配制一系列不同含量的标准溶液,以不含被测组分的空白溶液为参比,在相同条件下测定标准溶液的吸光度,绘制吸光度-浓度曲线。这种曲线即称为校准曲线。在相同条件下测定未知试样的吸光度,从校准曲线上就可以找到与之对应的未知试样的浓度。在建立一个方法时,首先要确定符合Lambert-Beer定律的浓度范围,即线性范围,定量测定一般在线性范围内进行。
(2)比较法 在相同条件下测定试样溶液和某一浓度的标准溶液的吸光度![]() 和
和![]() ,由标准溶液的浓度
,由标准溶液的浓度![]()
![]() ,
,![]() ,
,![]() (3.20)
(3.20)
这种方法比较简便,但只有在测定的浓度范围内溶液完全遵守Lambert-Beer定律,并且![]() 和
和![]() 很接近时,才能得到较为准确的结果。
很接近时,才能得到较为准确的结果。
2.多组分定量方法
根据吸光度具有加和性的特点,在同一试样中可以测定两个以上的组分。假设试样中含有![]() 两种组合,在一定条件下将它们转化为有色化合物,分别绘制其吸收光谱,会出现3种情况,如图3.14所示。图3.14(a)中两组分互不干扰,可分别在
两种组合,在一定条件下将它们转化为有色化合物,分别绘制其吸收光谱,会出现3种情况,如图3.14所示。图3.14(a)中两组分互不干扰,可分别在![]() 和
和![]() 处测量溶液的吸光度。图3.14(b)中组分x对组分y的光度测定有干扰,但组分y对x无干扰。这时可以先在
处测量溶液的吸光度。图3.14(b)中组分x对组分y的光度测定有干扰,但组分y对x无干扰。这时可以先在![]() 处测量溶液的吸光度
处测量溶液的吸光度![]() ,并求得x组分的浓度,然后再在
,并求得x组分的浓度,然后再在![]() 处测量溶液的吸光度
处测量溶液的吸光度![]() 和纯组分x和y的
和纯组分x和y的![]() 和
和![]() 值,根据吸光度的加和性原则,可列出下式
值,根据吸光度的加和性原则,可列出下式
![]() (3.21)
(3.21)
由式(3.21)即能求得组分y的浓度![]()
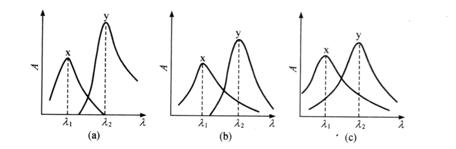
图3.14 多组分的吸收光谱
(a)组分x和y互不干扰 (b)组分x干扰y(y组分不干扰x) (c)组分x和y互相干扰
图3.14(c)表明两组分彼此互相干扰,这时首先在![]() 处测定混合物吸光度
处测定混合物吸光度![]() 和纯组分的x及y的
和纯组分的x及y的![]() 和
和![]() 。然后在
。然后在![]() 处测定混合物吸光度
处测定混合物吸光度![]() 和纯组分的
和纯组分的![]() 和
和![]() 。根据吸光度的加和性原则,可列出方程式
。根据吸光度的加和性原则,可列出方程式
![]()
![]() (3.22)
(3.22)
式中![]() 和
和![]() 均由己知浓度x及y的纯溶液测得。试液的
均由己知浓度x及y的纯溶液测得。试液的![]() 和
和![]() 由实验测得,
由实验测得,![]() 和
和![]() 便可通过解联立方程式求得。对于更复杂的多组分体系,可用计算机处理测定的数据。
便可通过解联立方程式求得。对于更复杂的多组分体系,可用计算机处理测定的数据。
3.双波长法
当吸收光谱相互重叠的两种组分共存时,利用双波长可对单个组分进行测定或同时对两个组分进行测定。如图3.15所示,当a、b两组分共存时,如要测定组分b的含量,组分a的干扰可通过选择具有对a组分等吸收的两个波长![]() 和
和![]() 加以消除。以
加以消除。以![]() 为参比波长,
为参比波长,![]() 为测定波长,对混合液进行测定,可得到如下方程式
为测定波长,对混合液进行测定,可得到如下方程式
![]()
![]() (3.23)
(3.23)
式中![]() 和
和![]() 是在波长
是在波长![]() 和
和![]() 下的背景吸收。当两个波长相距较近时,可认为背景吸收相等,因此通过试样吸收池的两个波长的光的吸光度差值为
下的背景吸收。当两个波长相距较近时,可认为背景吸收相等,因此通过试样吸收池的两个波长的光的吸光度差值为
![]() (3.24)
(3.24)
由于干扰组分a在![]() 和
和![]() 处具有等吸收,即
处具有等吸收,即![]() ,因此上式为
,因此上式为
![]() (3.25)
(3.25)
对于被测组分b来说,(![]() )为一定值,吸收池厚度
)为一定值,吸收池厚度![]() 也是固定的,所以
也是固定的,所以![]() 与组分b的浓度
与组分b的浓度![]() 呈正比。同样,适当选择组分b具有等吸收的两个波长,也可以对组分a进行定量测定,这种方法称为双波长等吸收点法。
呈正比。同样,适当选择组分b具有等吸收的两个波长,也可以对组分a进行定量测定,这种方法称为双波长等吸收点法。
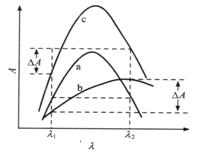
图3.15 双波长法测定示意图
a、b. 分别为组分a、b 的吸收曲线 c.两组分混合后的吸收曲线
当干扰组分的吸收曲线在测量的波长范围内无吸收峰时,等吸收点法就无法应用,这时可采用系数倍率法进行测定,并采用具有双波长功能的分光光度计来完成。假设被测组分为![]() 。干扰组分为
。干扰组分为![]() ,选择两个波长
,选择两个波长![]() 和
和![]() ,使
,使![]() 和
和![]() 的两束光分别通过吸收池,得到两个吸光度值A1和A2,然后由函数放大器分别放在
的两束光分别通过吸收池,得到两个吸光度值A1和A2,然后由函数放大器分别放在![]() 和
和![]() 倍,由此得到差示信号
倍,由此得到差示信号![]()
![]()
式中![]() 和
和![]() 分别是两组分混合物在波长
分别是两组分混合物在波长![]() 和
和![]() 处的吸光度,即
处的吸光度,即
![]()
则
![]()
调节信号放大器,选取![]() 和
和![]() ,使之满足
,使之满足
![]()
此时组分![]() 在
在![]() 和
和![]() 处显示等同信号,即
处显示等同信号,即![]() ,在此条件下
,在此条件下
![]() (3.26)
(3.26)
这样,差示信号![]() 就只与被测组分x的浓度
就只与被测组分x的浓度![]() 有关,因而有可能测定出混合物中组分x的含量。
有关,因而有可能测定出混合物中组分x的含量。
4.示差分光光度法
示差分光光度法(differential spectrophotometry)有4种类型,即高吸光度示差法、低吸光度示差法、最精密示差测量法和全示差光度测量法。应用较广是测定高含量组分的高吸光度示差法。
高吸光度示差法是采用浓度比试样含量稍低的己知浓度的标准溶液作为参比溶液。如果标准溶液浓度为![]() ,待测试样浓度为
,待测试样浓度为![]() ,而且
,而且![]() >
>![]() 。根据朗伯-比尔定律
。根据朗伯-比尔定律
![]()
![]() (3.27)
(3.27)
测定时先用经试样浓度稍小的标准溶液,加入各种试剂后作为参比,调节其透射比为100%,即吸光度为零,然后测量试样溶液的吸光度。这时的吸光度实际上是两者之差![]() ,它与两者浓度差
,它与两者浓度差![]() 呈正比,且处在正常的读数范围(图3.16)。
呈正比,且处在正常的读数范围(图3.16)。
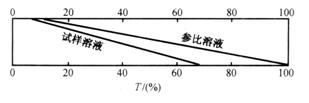
图3.16 高吸光度示差法测定原理示意图
以![]() 与
与![]() 作校准曲线,根据测得
作校准曲线,根据测得![]() 查得相应的
查得相应的![]() ,则
,则![]() 。
。
由于用已知浓度的标准溶液作参比,如果该参比溶液的透射比为10%,现调至100%,就意味着将仪器透射比标尺扩展了10倍。如待测试液的透射比原是5%,用示差光度法测量时将是50%。另一方面,在示差光度法中即使![]() 很小,如果测量误差为dc,固然dc/
很小,如果测量误差为dc,固然dc/![]() 会相当大,但最后测定结果的相对误差是
会相当大,但最后测定结果的相对误差是![]() ,
,![]() 较大而非常准确,所以测定结果的准确度仍然将很高。
较大而非常准确,所以测定结果的准确度仍然将很高。
5.导数分光光度法
导数分光光度法(derivative
spectrophotometry)是解决干扰物质与被测物质的吸收光谱重叠,消除胶体和悬浮物散射影响和背景吸收,提高光谱分辨率的一种技术。将Lambert-Beer定律![]() 对波长
对波长![]() 进行
进行![]() 次求导,得到
次求导,得到
![]() (3.28)
(3.28)
由式(3.28)可知,吸光度的导数值仍与吸光物质的浓度呈线性关系,借此可以进行定量分析。
图3.17为物质的吸收光谱(零阶导数光谱)和它的1~4阶导数光谱图。由图可见,随着导数的阶次增加,谱带变得更加尖锐,分辨率提高。
在用导数光谱进行定量分析时,需要对扫描出的导数光谱进行测量以获得导数值。常用的测量方法有3种,如图3.18所示。
(1)正切法 画一条直线正切于两个邻近的极大或极小,然后测量中间极值至切线的距离![]() 。这种方法可用于线性背景干扰的试样的测定。
。这种方法可用于线性背景干扰的试样的测定。
(2)峰谷法 在多组分的定量分析中多采用两个相邻极值(极大或极小)间的距离(图3.18中的![]() 和
和![]() )作为导数值。
)作为导数值。
(3)峰零法 极值到零线之间的垂直距离![]() 也可以作为导数值。这种方法适用于信号对称于横坐标的较高阶导数的求值。
也可以作为导数值。这种方法适用于信号对称于横坐标的较高阶导数的求值。
由于导数光谱的灵敏度高、再现性好、噪声低、分辨率高等优点,一些物质,如核糖核酸酶A、过氧化氢酶、纤维朊原、细胞色素c等的高阶导数光谱显出它们特征的精细结构,称为“指纹”光谱,可用于这些物质的鉴定和纯度检验。
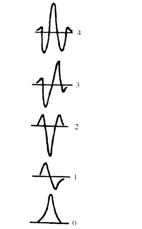
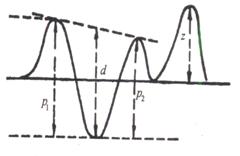

图3.17 物质的吸收曲线及其1~ 4阶导数光谱
应用光度法测定络合物组成的方法有多种,这里介绍两种常用的方法。
(1)摩尔比法(mole ratio method)(又称饱和法) 它是根据金属离子M在与配位体R反应过程中被饱和的原则来测定络合物组成的。
设配合反应为 M + nR = MRn
若M与R均不干扰MR![]() 的吸收,且其分析浓度分别是
的吸收,且其分析浓度分别是![]() 、
、![]() ,那么固定金属离子M的浓度,改变配位体R的浓度,可得到一系列
,那么固定金属离子M的浓度,改变配位体R的浓度,可得到一系列![]() /
/![]() 不同的溶液。在适宜波长下测定各溶液的吸光度,然后以吸光度
不同的溶液。在适宜波长下测定各溶液的吸光度,然后以吸光度![]() 对
对![]() /
/![]() 图(图3.19)。当加入的配位体R还没有使M定量转化为MR
图(图3.19)。当加入的配位体R还没有使M定量转化为MR![]() 并稍有了过量时,曲线便出现转折;加入的R继续过量,曲线便成水平直线。转折点所对应的摩尔比数便是络合物的组成比。若络合物较稳定,则转折点明显;反之,则不明显,这时可用外推法求得两直线的交点。交点对应的
并稍有了过量时,曲线便出现转折;加入的R继续过量,曲线便成水平直线。转折点所对应的摩尔比数便是络合物的组成比。若络合物较稳定,则转折点明显;反之,则不明显,这时可用外推法求得两直线的交点。交点对应的![]() 即是
即是![]() 。
。
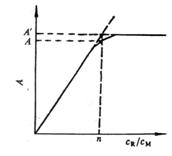
图3.19 摩尔比法
(2)等摩尔系列法(equimolar
series method)(又称Job法)设络合反应为
M + nR = MRn
设cM和cR分别为溶液中M与R物质的量浓度,配制一系列溶液,保持cM +cR=c(c恒定)。改变cM和cR的相对比值MRn,在的最大吸收波长下测定各溶液的吸光度![]() 。当
。当![]() 达到最大时,即浓度MRn最大,该溶液中cM/cR比值即为络合物的组合比。如以吸光度
达到最大时,即浓度MRn最大,该溶液中cM/cR比值即为络合物的组合比。如以吸光度![]() 为纵坐标值cM/c为横坐标作图,即绘出等摩尔系列法曲线(图3.20)。
为纵坐标值cM/c为横坐标作图,即绘出等摩尔系列法曲线(图3.20)。
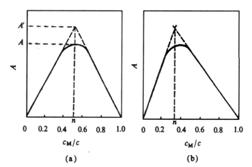
图3.20 等摩尔系列法
图中,两曲线外推的交点所对应的cM/c 即是配合物的组成M与R的摩尔比(n值)。
该法适用于溶液中只形成一种离解度小的、配合比低的络合物组成的测定。
光度法是测定分析化学中应用的指示剂或显色剂离解常数的常用方法,因为它们大多是有机弱酸或弱碱,只要它们的酸色形式和碱色形式的吸收曲线不重叠。该法特别适用于溶解度较小的弱酸或弱碱。
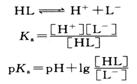 现以一元弱酸HL为例,在溶液中有如下平衡关系
现以一元弱酸HL为例,在溶液中有如下平衡关系
其离解常数
或 (3.29)
从式(3.29)可知,只要在某一确定的pH下,知道[HL]与[L-]的比值,就可以计算pK![]() 。HL是L-互为共轭酸碱,它们的平衡浓度之和等于弱酸HL的分析浓度
。HL是L-互为共轭酸碱,它们的平衡浓度之和等于弱酸HL的分析浓度![]() 。只要两者都遵从朗伯-比尔定律,就可以通过测定溶液的吸光度求得[HL]和[L-]的比值。具体做法是:配制
。只要两者都遵从朗伯-比尔定律,就可以通过测定溶液的吸光度求得[HL]和[L-]的比值。具体做法是:配制![]() 个浓度
个浓度![]() 相等而pH不同的HL溶液,在某一确定的波长下,用
相等而pH不同的HL溶液,在某一确定的波长下,用![]() ,并用酸度计测量各溶液的pH。各溶液的吸光度为
,并用酸度计测量各溶液的pH。各溶液的吸光度为
![]() (3.30)
(3.30)
![]()
在高酸度介质中,可以认为溶液中该酸只以HL型体存在,仍在以上确定的波长下测定吸光度,则
![]()
![]() (3.31)
(3.31)
而在碱性介质中,可以认为该酸主要以L-型体存在,这时依然在以上波长下测量吸光度,则
![]()
![]() (3.32)
(3.32)
将式(3.31)、(3.32)代入式(3.30),整理后,得
![]()
或
![]() (3.33)
(3.33)
上式是用光度法测定一元弱酸离解常数的基本关系式。式中![]() 、
、![]() 分别为弱酸定量地以HL、L-型体存在时溶液的吸光度,该两值是不变的;
分别为弱酸定量地以HL、L-型体存在时溶液的吸光度,该两值是不变的;![]() 为某一确定pH时溶液的吸光度。上述各值均可由实验测得,将测定的数据代入式(3.33)就可算出pK
为某一确定pH时溶液的吸光度。上述各值均可由实验测得,将测定的数据代入式(3.33)就可算出pK![]() ,然后取其平均值;也可以将实验数据采用线性拟合法或作图法求出pK
,然后取其平均值;也可以将实验数据采用线性拟合法或作图法求出pK![]() 。
。
[例3.8] 紫外-可见吸收光谱法测定防腐剂。
|
分析样品 |
饮料、食品。 |
|
分析项目 |
苯甲酸、山梨酸(防腐剂)。 |
|
分析方法 |
用乙醚萃取法将防腐剂从饮料(食品)中提取出来,然后用紫外光谱法定性和定量。 |
|
分析条件 |
(1)分离样品中的防腐剂*:用C2H5OC2H5萃取后,在容量瓶中定容。 (2)防腐剂的定性鉴定:经提纯稀释后C2H5OC2H5萃取液(或水溶液)用 (3)防腐剂的定量测定: 配制苯甲酸(或山梨酸)的标准系列溶液,并定容。 用 用定性鉴定后的样品的C2H5OC2H5萃取液(或稀释液)按上述方法测定吸光度。 |
|
分析结果 |
(1)定性 根据试样的吸收峰,吸收强度以及它与苯甲酸(228nm和270nm处分别有K吸收带和B吸收带)和山梨酸(在255nm处有吸收带)标准样吸收光谱对照,以确定防腐剂种类。 (2)定量 采用最小二乘法处理标准溶液的浓度和吸光度数据,以求得浓度与吸光度之间的线性回归方程,并根据线性回归方程计算样品中防腐剂的含量。 |
*如果测定试样中无干扰组分,则无须分离,可直接测定。
[例3.9] 紫外-可见分光光度法测定维生素A。
|
分析样品 |
新鲜鸡肝 |
|
分析项目 |
维生素A
(脂溶性质淡黄色晶体) |
|
分析方法 |
用乙醚将样品中的脂肪及维生素A提取出来,除去溶剂后进行皂化以除去脂肪,皂化后再进行萃取以便将维生素A转入有机相中,然后经柱色谱除去干扰物质,最后用紫外分光光度计进行吸光度测定,用标准曲线法求出样品中维生素A的含量。 |
|
分析条件 |
(1)提取与皂化 将样品洗净、处理后用C2H5OC2H5振荡提取,将提取液中的C2H5OC2H5蒸干后,加入80%KOH溶液、乙醇和焦性没食子酸,置于(83±1)℃的水浴中回流皂化。 (2)萃取、洗涤、浓缩 将皂化后的混合液在一定条件下用C2H5OC2H5萃取。然后加KOH于C2H5OC2H5提取液中,弃去下层碱液,再用水洗涤,直至洗液与酚酞无颜色反应为止,弃去水层。 将上述C2H5OC2H5提取液经过无水Na2SO4滤入锥形瓶,置于水浴上,把C2H5OC2H5蒸干后,加入石油醚溶解锥形瓶中内容物,备用。 (3)色谱分离 将上述石油醚样品溶液移入Al2O3及无水Na2SO4的色谱柱中,用不同比例的乙醚-石油醚洗脱液进行梯度洗脱,在12%洗脱液前后洗出的第一个黄色色谱带为 (4)制备维生素A标准系列溶液。 (5)测定 用紫外分光光度计, |
|
分析结果 |
(1)绘制标准曲线(吸光度 (2)根据试样溶液的吸光度在标准曲线上查出相应的质量浓度 (3)按下式计算样品中维生素A的质量分数:
式中,v为测定时容量瓶的体积(单位为mL); |
思考题与习题
3.1 电子跃迁有哪几种类型?哪些类型的跃迁能在紫外-可见吸收光谱中反映出来?
3.2 朗伯-比尔定律的物理意义是什么?偏离朗伯-比尔定律的原因主要有哪些?
3.3 吸光度与透射率有什么关系?物质溶液的颜色与光的吸收有什么关系?
3.4 什么是发色团及助色团?举例说明。
3.5 下列化合物各具有几种类型的价电子?在紫外光照射下发生哪几种类型的电子跃迁?
乙烷
磺乙烷
丙酮
丁二烯 苯乙烯 苯乙酮
3.6 试比较下列化合物,指出哪个吸收光的波长最长?哪个最短?为什么?
3.7
某苦味酸胺试样![]() =4.13)
=4.13)
3.8
称取钢样![]() ,用水稀至刻度,摇匀。于520 nm处
,用水稀至刻度,摇匀。于520 nm处![]() =2.3×
=2.3×
3.9 己知一物质在它的最大吸收波长处的摩尔吸收系数![]() 为1.4×
为1.4×
3.10 .K2CrO4的碱性溶液在372nm处有最大吸收,若碱性K2CrO4溶液的浓度为3.00×10-5mol·L-1,吸收池厚度为1nm,在此波长下测得透射率是71.6%,计算:(1)该溶液的吸光度;(2)摩尔吸收系数;(3)若吸收池厚度为
3.11
苯胺在![]() 为280nm处的
为280nm处的![]() 为
为
3.12某组分A溶液的浓度为5.00×10-4mol·L-1,在